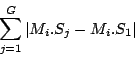Overview GRIL locates the breakpoints of inversions and rearrangements in DNA sequences. The breakpoints located by GRIL represent the boundaries of collinear regions of sequence called Locally Collinear Blocks (LCBs). Each locally collinear block may contain non-homologous sequence regions but may not contain significant rearrangements of homologous sequence. In GRIL, significance of a particular homologous region is determined by user-specified parameters such as the length of the surrounding collinear region. GRIL uses three basic steps to locate inversions and rearrangements. First, a string matching algorithm locates regions of sequence identity. Second, some of the matching regions found in the first step are filtered out based on user specified criteria. Finally, the remaining regions of sequence identity are organized into Locally Collinear Blocks (LCBs).
Step 1: Locating Regions of Sequence Identity The first step of GRIL involves locating regions of sequence identity on which it bases its inference of collinear regions. The particular string matching algorithm implemented in GRIL locates maximal unique matches (MUMs) present in each sequence under study. A maximal unique match is an identical region in some set of sequences that could not possibly be extended to include a larger region without including a mismatching character. A MUM is unique because the complete character sequence in the region it covers is not repeated anywhere else in the DNA sequence. In other words, the DNA sequence in the region covered by a MUM is present exactly once in each genome. Formally we define a MUM as the tuple 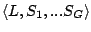 where where 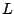 is the length of the MUM, is the length of the MUM, 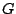 is the number of sequences input to GRIL, and is the number of sequences input to GRIL, and 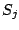 is the left-end position of the MUM in sequence is the left-end position of the MUM in sequence 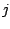 . The match finding step of GRIL outputs a set of MUMs . The match finding step of GRIL outputs a set of MUMs 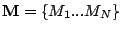 . The . The 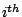 MUM in MUM in 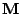 is referred to as is referred to as 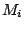 . To refer to the length of we use the notation . To refer to the length of we use the notation 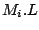 and similarly, we refer to the left end of in sequence using the notation and similarly, we refer to the left end of in sequence using the notation 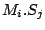 . Finally, if MUM includes a region in the reverse complement orientation in sequence , we define the sign of to be negative. For a further description of MUMs, see Alignment of Whole Genomes (Delcher et. al 1999). . Finally, if MUM includes a region in the reverse complement orientation in sequence , we define the sign of to be negative. For a further description of MUMs, see Alignment of Whole Genomes (Delcher et. al 1999). To locate MUMs in several sequences simultaneously, GRIL implements an extension of traditional seed-and-extend style string matching to multiple sequences. First, a data structure called a Sorted Mer List (SML) is constructed for each sequence. The SMLs are then used to identify candidate MUM seed matches. If a candidate MUM seed is not part of an existing MUM, the seed is extended and added to a list of known MUMs.
Step 2: Filtering Matches Once a set of MUMs has been identified, GRIL removes some MUMs from the set based on user-specified filtering criteria. There are four criteria that GRIL can use to filter MUMs at this stage: GRIL applies the filters in the following order: minimum length threshold, maximum generalized offset threshold, minimum identity threshold, minimum range threshold.
Step 3: Determining Locally Collinear Blocks (LCBs) In this final step, GRIL determines Locally Collinear Blocks based on the set of MUMs remaining after filtering has been applied. The output of this step is a set of LCBs. An LCB can be defined formally as a collinear region was defined in the previous section, specifically 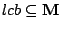 where is the MUM in the LCB. The MUMs that constitute an LCB must satisfy a total ordering property such that where is the MUM in the LCB. The MUMs that constitute an LCB must satisfy a total ordering property such that 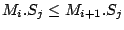 holds for all i, holds for all i, 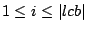 and all , and all , 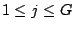 . . GRIL uses a standard breakpoint determination algorithm to partition the set of filtered MUMs into a set of LCBs. First, GRIL orders the MUMs on 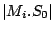 . Next, a monotonically increasing label between 1 and . Next, a monotonically increasing label between 1 and 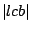 is assigned to each MUM corresponding to the index of the MUM in the ordering on . We will refer to the label of the MUM as is assigned to each MUM corresponding to the index of the MUM in the ordering on . We will refer to the label of the MUM as 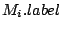 . Note that . Note that 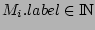 . Next, the set of MUMs is repeatedly reordered based on . Next, the set of MUMs is repeatedly reordered based on 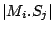 for for 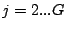 . After each reordering, the set of MUMs are examined for breakpoints. A breakpoint exists between and . After each reordering, the set of MUMs are examined for breakpoints. A breakpoint exists between and 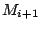 if if 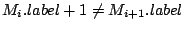 and both and are in the forward orientation, or if and both and are in the forward orientation, or if 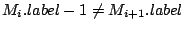 and both and are in the reverse complement orientation. A breakpoint also exists if is in a different orientation than in sequence , e.g. the sign of is different than the sign of and both and are in the reverse complement orientation. A breakpoint also exists if is in a different orientation than in sequence , e.g. the sign of is different than the sign of 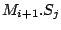 . . The recorded set of breakpoints defines a partitioning of the set of filtered MUMs into a set of LCBs.
Memory Requirements The sorted mer list construction phase is the most memory intensive component of the GRIL algorithm. To construct an SML, GRIL requires approximately 16 bytes of memory per base of the input sequence. Since each SML is written to disk after construction GRIL's memory requirements are proportional to the length of the longest sequence. For example, detecting rearrangements in 7 bacterial genomes, the largest of which is 5.5MB, would require approximately 88MB of memory.
|